RESEARCH
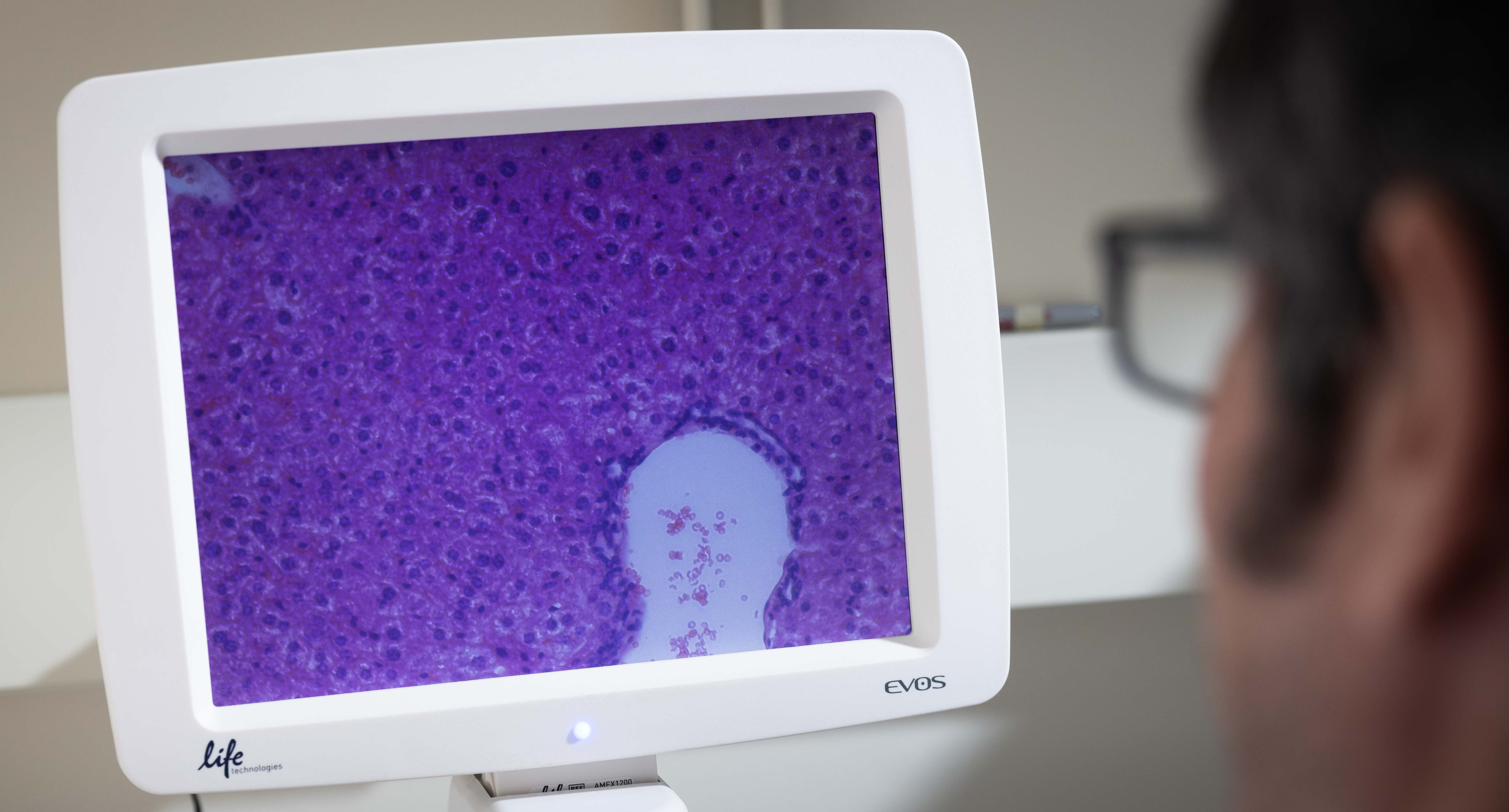
The Hormel Institute Scientist Susan Hafenstein, PhD, Receives $85K Grant to Study Hepatitis E
This support will enable the Hafenstein lab to use cryo-electron microscopy (cryoEM) and tomography (cryoET) to study the structure and functions of the hepatitis E virus (HEV), which could help lead to the development of new treatment and preventative measures against HEV hepatitis.
MBiC
Austin Port Authority, Legislators, and The Hormel Institute Members Hold Press Conference for MBiC at State Capitol
“[This isn’t] just an Austin project. This is with Mayo Clinic, University of Minnesota, and this is going to impact the state, the nation, and the world, because they are leading in cancer research and treatment,” said Senator Gene Dornink.

Minnesota Bioimaging Center
MBiC is a phased project that will build innovation space for cutting-edge bioimaging research capabilities needed to accelerate scientific discoveries in Minnesota and the Midwest region, educate and support the next generation of researchers, and create a positive economic impact for our communities.