No Longer a 'Secret': Gov. Tim Walz Praises MBiC Project at The Hormel Institute Visit
Gov. Walz shared that he used to refer to The Hormel Institute as Minnesota’s “best-kept secret,” but no longer finds that to be the case, acknowledging the vision of MBiC aligns with many statewide priorities for the future.
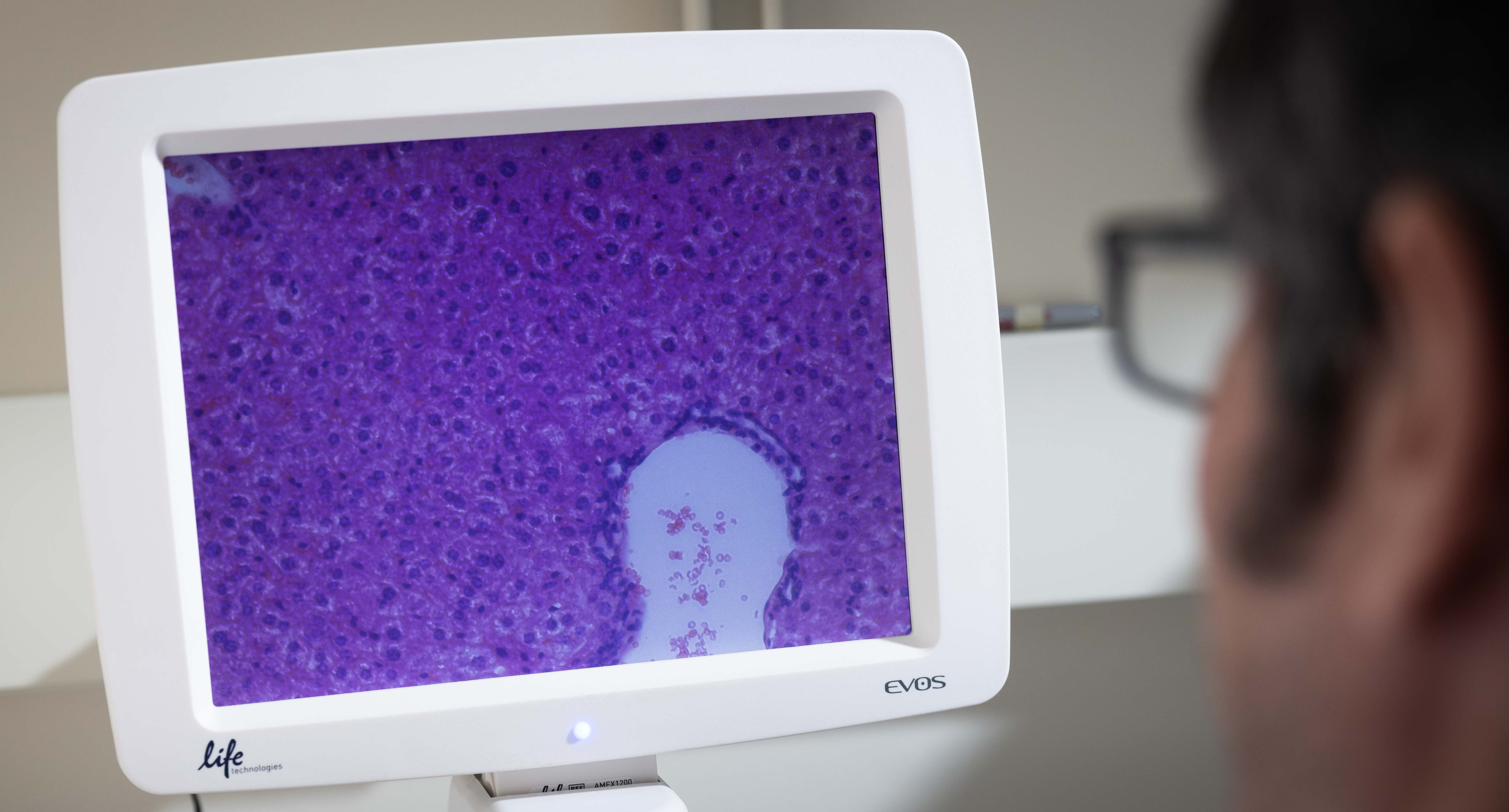
Study Published Could Offer Solution For Reawakening Immune System Responses to Inhibit Cancer Progression
Robert Clarke, PhD, Executive Director and Professor, and Lu Jin, MS, Researcher at The Hormel Institute, University of Minnesota, have co-authored a paper recently published in the journal ACS Nano.

Minnesota Bioimaging Center
MBiC is a phased project that will build innovation space for cutting-edge bioimaging research capabilities needed to accelerate scientific discoveries in Minnesota and the Midwest region, educate and support the next generation of researchers, and create a positive economic impact for our communities.